Determine P-site offsets for ribosome profiling data¶
In this tutorial we:
determine the P-site offset to use with a ribosome profiling dataset.
use these offsets as a mapping rule to map read alignments from that dataset to their P-sites, enabling visualization of translation
Here, we presume that ribosome-protected fragments were produced using an unbiased nuclease (e.g. RNase I, NOT microccal nuclease). that trims the ribosome-protected footprints to the edges of the ribosome.
We will use the Demo dataset.
The following sections appear in this document:
Strategy¶
Following [IGNW09], an effective way to map P-sites is to use peaks of ribosome density that occur over start codons.
The ribosomal P-site is somewhere internal to the ribosome-protected footprint generated by ribosome profiling. Therefore, when we map read alignments of footprints to their 5’ ends, we would expect to see the start codon peak at some fixed distance upstream of the start codon in a metagene analysis of the data. The distance between the observed start codon peak and the annotated start codon corresponds to the offset:
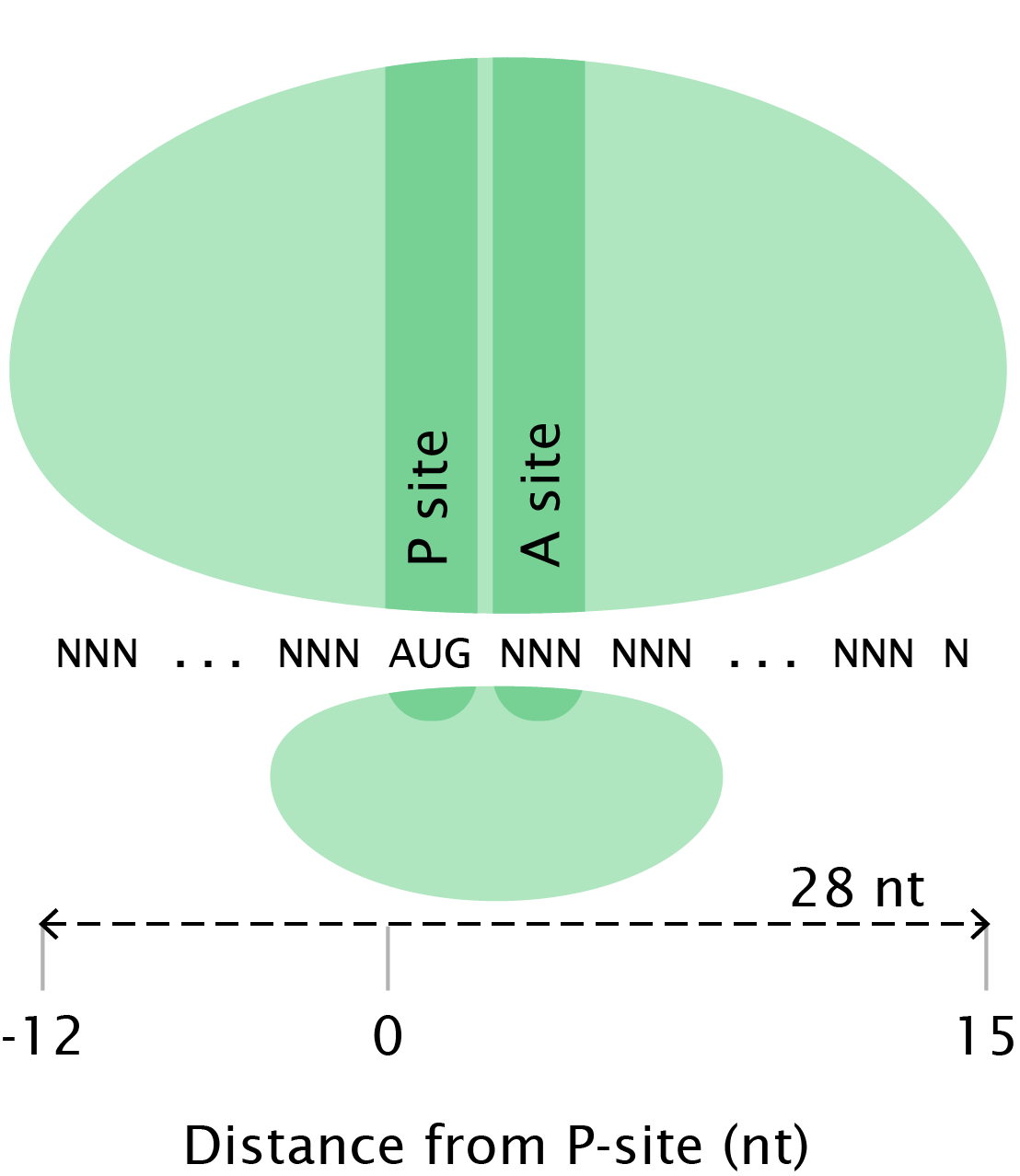
A ribosome containing a footprint after digestion. The P-site offset is the distance from the 5’ end of the read to the ribosomal P-site (in this case, 12 nt). After [IGNW09].¶
This yields the following strategy:
Separate footprints into classes based upon their lengths
For each length:
Perform a metagene analysis at the start codon, in which the footprints are mapped to their 5’ ends.
Measure the distance between the highest peak 5’ of the start codon and the start codon. Assuming this peak is the initiation peak, this distance is the offset to use for reads of this length:
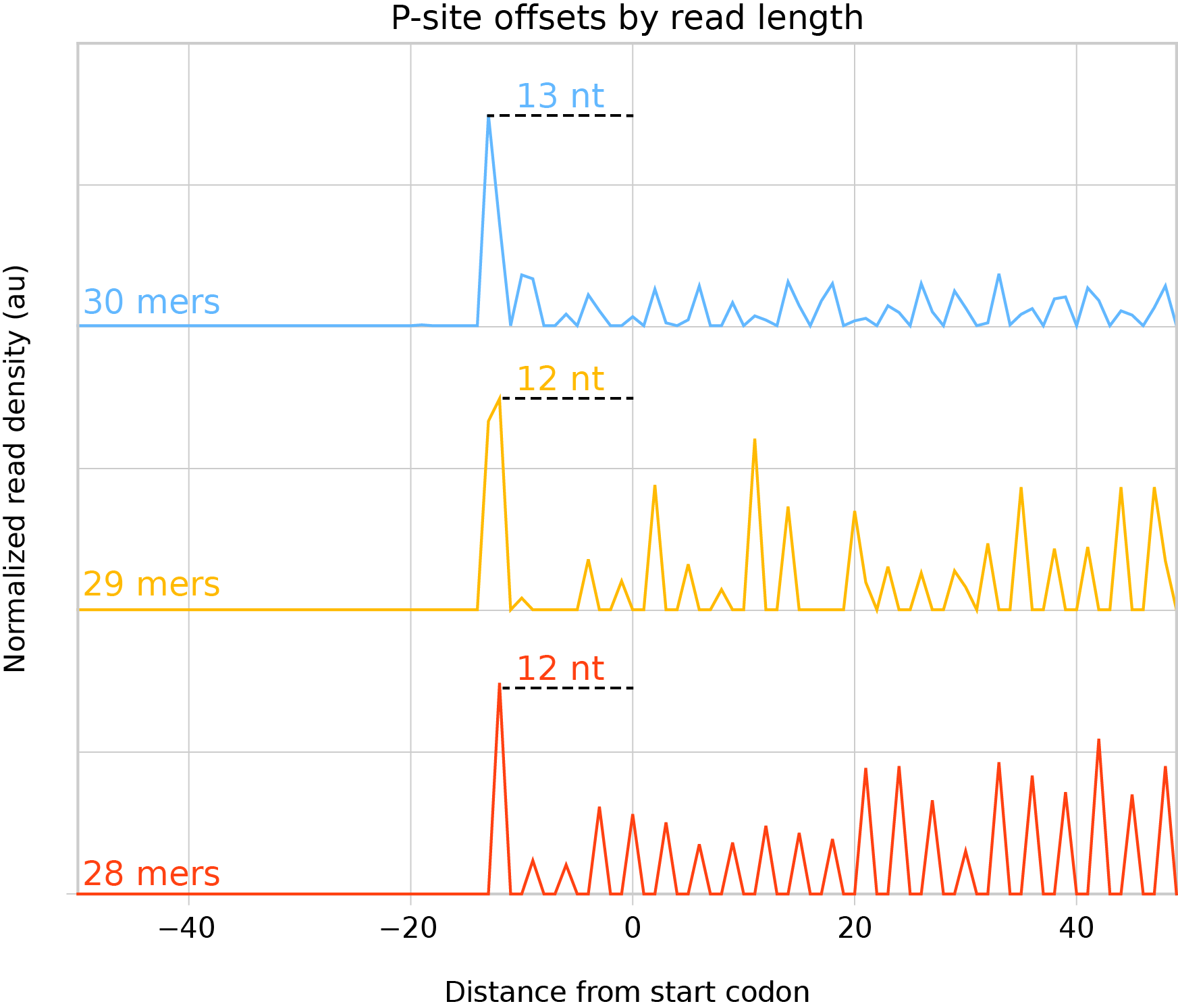
Metagene analysis of individual read lengths mapped to their 5’ ends to discover P-site offsets¶
Manually inspect offsets to make sure they seem reasonable
Check results by perform a metagene analysis around the start codon, this time using the P-site offsets we determined. Results should resemble the image below:
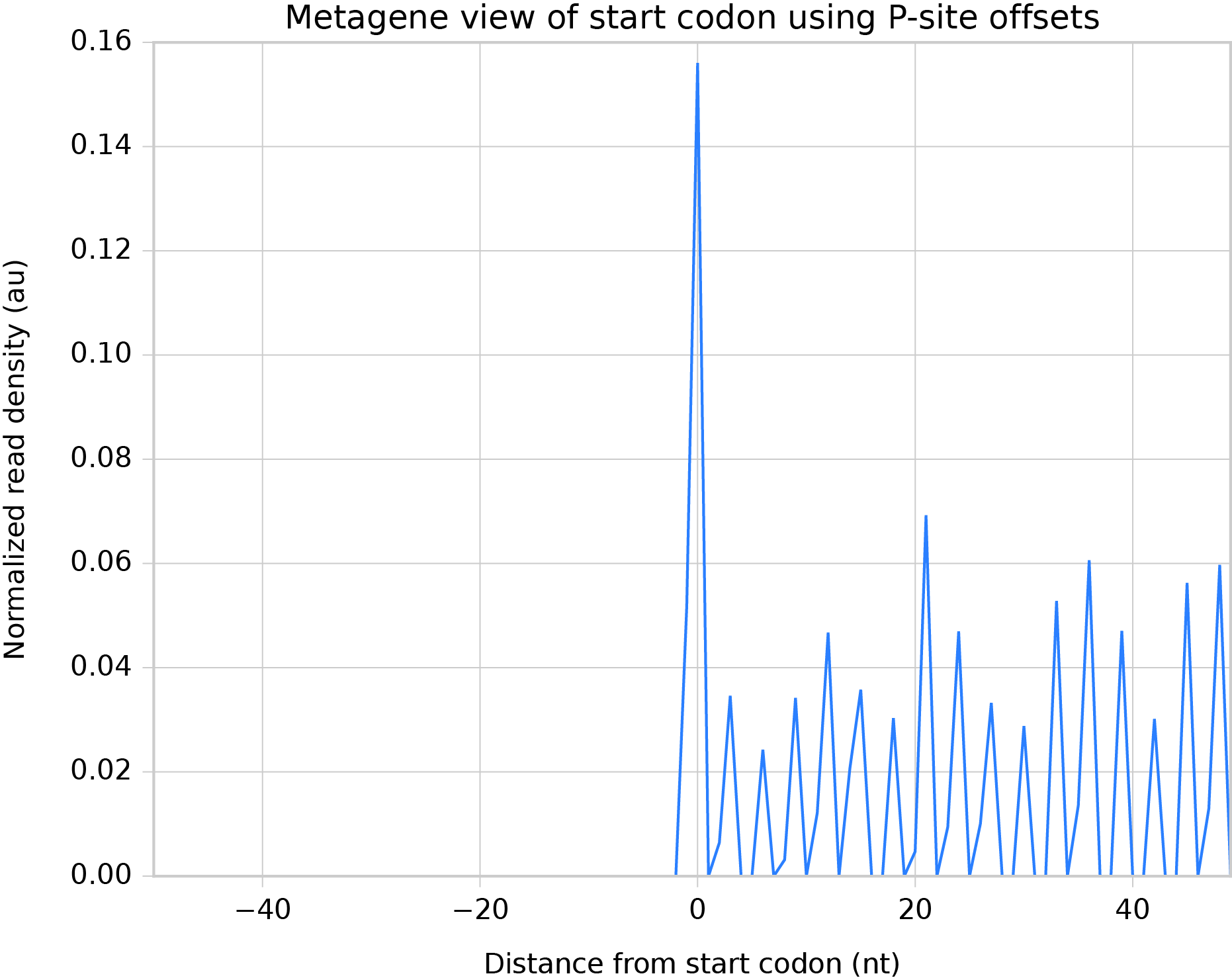
Metagene analysis surrounding start codon, with P-site offsets applied to read alignments¶
Determining P-site offsets using the psite script¶
The strategy above is implemented by psite, which can be executed from
the terminal.
Because psite internally performs metagene analysis, we need
to use a file of maximal spanning windows
produced by the metagene script. The command call to metagene
is included below, and explained in detal in Performing metagene analyses.
From the terminal:
# generate metagene `roi` file. See `metagene` documentation for details
$ metagene generate merlin_orfs \
--landmark cds_start \
--annotation_files merlin_orfs.gtf
# run the psite script
# We ignore reads shorter than 29 nucleotides or longer than 35-
# there should be few of these, and it saves psite from doing
# unnecessary analyses
$ psite merlin_orfs_rois.txt SRR609197_riboprofile \
--min_length 29 \
--max_length 35 \
--require_upstream \
--count_files SRR609197_riboprofile_5hr_rep1.bam
For most users, two of the output files are of interest:
A graphic (in this example,
SRR609197_riboprofile_p_offsets.png), showing the metagene profile for each read length: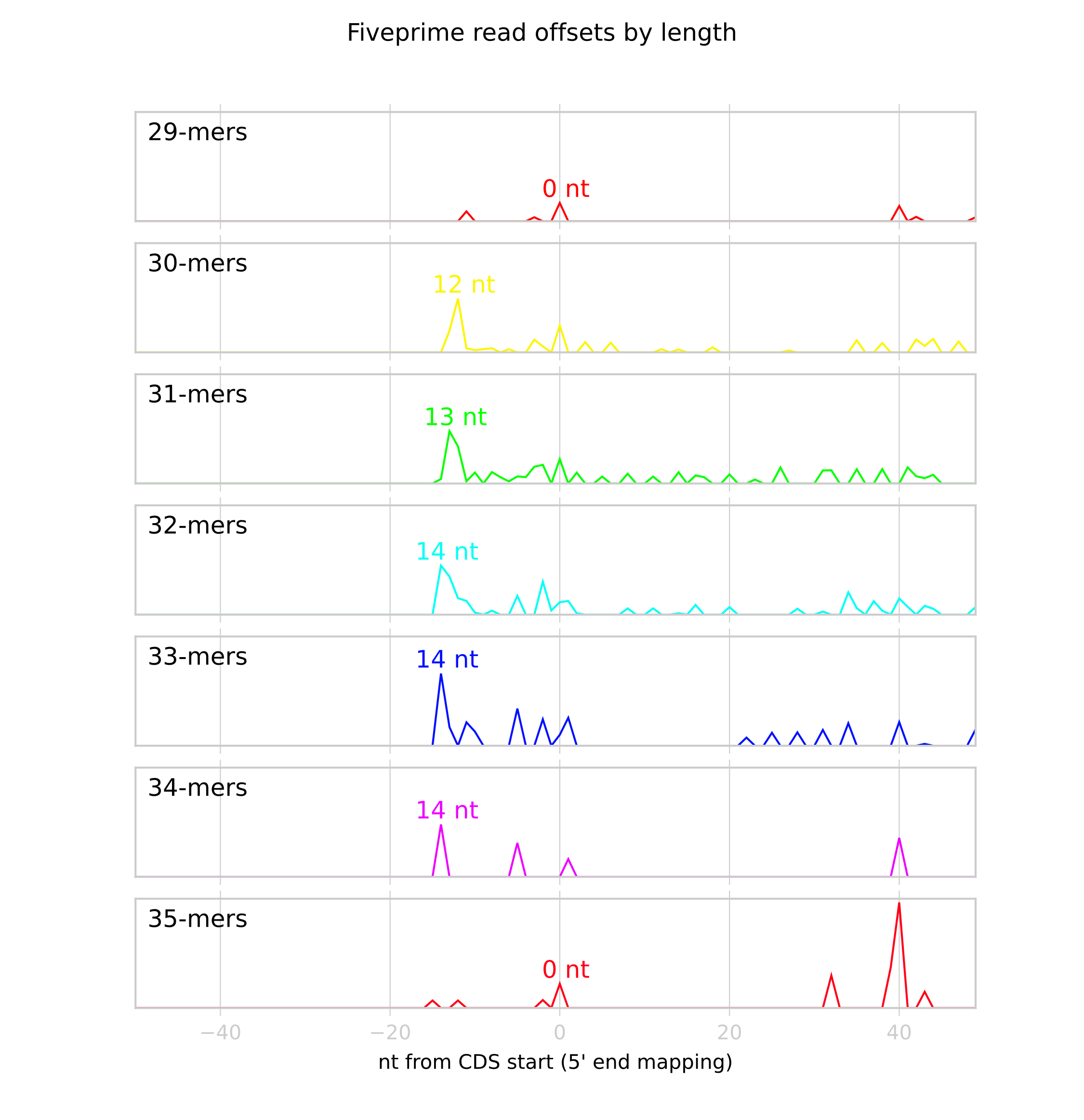
From this image we can see that there are few 29- and 35-mers, so their P-site mapping is likely to be off. We’ll adjust these manually below.
A two-column text file (in this example,
SRR609197_riboprofile_p_offsets.txt), in which the first column is a read length and the second, the corresponding P-site offset from the 5’ end of the read:#length p_offset 29 0 30 12 31 13 32 14 33 14 34 14 35 0 default 13
As in the graphical output, the values for 29 and 35 appear to be off. We will edit this file in a text editor, and set the offset to 12 for 29-mers, and 14 for 35-mers. We’ll also set the default to 14, the most common value. This gives the following table:
#length p_offset 29 12 30 12 31 13 32 14 33 14 34 14 35 14 default 14
If the output looks blank for one or more read lengths¶
This occurs in datasets in which there are few reads of any given length. In this case, there are a couple of options:
Raise the number of minimum counts required to be included in the analysis using the
--min_countsargumentEstimate the P-site offset from aggregate read counts at each position, instead of median normalized read density, using the
--aggregateargument.The columnwise aggregate measurement is potentially noisier than the columnwise median, but more sensitive to low read counts.
In either case, it is useful to add the --keep argument to keep some
intermediate files, in case we wish to aggregate or filter them manually:
# re-run the psite script using --aggregate
$ psite merlin_orfs_rois.txt merlin_orfs \
--min_length 29 \
--max_length 35 \
--require_upstream \
--count_files SRR609197_riboprofile_5hr_rep1.bam \
--aggregate --keep
To manipulate the data manually, load the appropriate data matrix from the
previous run (named SAMPLE_LENGTH_rawcounts.txt.gz):
>>> import numpy
>>> import matplotlib.pyplot as plt
>>> counts29 = numpy.loadtxt("merlin_orfs_29_rawcounts.txt.gz")
>>> profile29 = numpy.nansum(counts29,axis=0)
# assuming we used `--upstream 50 --downstream 50` in call to `metagene generate`
# change ranges below to match what you used
>>> x = numpy.arange(-50,50)
# estimate offset as highest peak upstream of start codon
>>> offset = 0 - x[profile29[x <= 0].argmax()]
# plot
>>> plt.plot(x,profile29,label="29 mers")
>>> plt.axvline(-offset,dashes=[2,2],label="%s nt offset" % offset)
# check estimate to see if it is reasonable
>>> plt.show()
Then, manually edit the text output accordingly.
Using the P-site offset in analyses¶
In command-line scripts¶
Command-line scripts in plastid use a common interface for
read mapping rules. To use the offsets generated by psite, use
the --fiveprime_variable mapping rule, and pass the text file made
by psite to the --offset parameter. For example, from the terminal:
$ some_script --fiveprime_variable \
--offset SRR609197_riboprofile_p_offsets_adjusted.txt [other arguments]
In interactive sessions¶
The mapping rule can be constructed by passing the offset file from psite to the
from_file()
method of VariableFivePrimeMapFactory:
>>> from plastid import *
>>> maprule = VariableFivePrimeMapFactory.from_file(open("merlin_orfs_p_offsets_adjusted.txt"))
>>> alignments = BAMGenomeArray("SRR609197_riboprofile_5hr_rep1.bam")
>>> alignments.set_mapping(maprule)
For alignments in bowtie-format use GenomeArray and
variable_five_prime_map():
>>> from plastid.util.io.filters import *
>>> from plastid.util.scriptlib.argparsers import _parse_variable_offset_file as pvof
>>> offset_dict = pvof(CommentReader(open("SRR609197_riboprofile_p_offsets_adjusted.txt")))
>>> alignments = GenomeArray()
>>> alignments.add_from_bowtie("some_file.bowtie",variable_five_prime_map,offset=offset_dict)
Pitfalls¶
This P-site mapping strategy requires pronounced initiation peaks in ribosome profiling data. If these are absent – which can happen under conditions of initiation shutdown (e.g., if the sample is under stress before lysis) – an alternative option is to use a stop codon peak (if present in the data) for mapping.
The simplest way to do this is to use the metagene script on reads
of separate lengths, again using fiveprime end mapping (--fiveprime
command-line argument passed to metagene), and manually inspecting
the output. For each read length, assign the offset to be the distance
between the stop codon and the peak (which should be tall, and followed
by a precipitous drop in ribosome density) immediately upstream of
the stop codon.
Is it necessary to separately estimate a P-site for each dataset?¶
Many experimentalists find that their technique is sufficiently consistent not to need to re-estimate P-site offsets for every dataset. Others are content to use offsets published in literature by other groups. Others more conservatively perform this analysis for every dataset. We strongly suggest performing this analysis at the very least:
when changing nuclease, buffer, or cloning conditions
when changing culture conditions (e.g. profiling under starvation, heat shock, viral infection, et c)
when ribosome profiling a new organism
See also¶
psitescript, for full documentation
metagenescript, for information on generating the ROI file used bypsitePerforming metagene analyses for an in-depth discussion of metagene analysis