Read mapping functions¶
Numerous sequencing assays encode interesting biology in various properties of read alignments withu nucleotide precision. For example:
ribosome profiling encodes the positions of ribosomal P-sites as a function of both read length and alignment coordinates ([IGNW09, ILW11])
Bisulfite sequencing encodes methylation status as C-to-T transitions within read alignments
DMS-Seq ([RZW+14]) and Pseudouridine profiling (:cite:) encode unstructured regions of RNA or sites of pseudouridine modification, respectively, in the 5’ termini fo their read aligments.
plastid uses configurable mapping functions to
decode the biology of interest from read alignments, and convert the decoded
data into arrays over regions of interest (such as an
array of the number of ribosomes at each nucleotide in a coding region).
This design enables plastid’s tools to operate on sequencing data from
virtually any NGS assay, provided the appropriate mapping function and
parameters.
Mapping functions are described in the following sections:
Mapping functions included in plastid¶
At present, the following mapping functions are provided, although users are encouraged to write their own mapping functions as needed. These include:
- Fiveprime end mapping
Each read alignment is mapped to its 5’ end, or at a fixed offset (in nucleotides) from its 5’ end
- Variable fiveprime end mapping
Each read alignment is mapped at a fixed distance from its 5’ end, where the distance is determined by the length of the read alignment.
This is used for ribosome profiling of yeast ([IGNW09]) and mammalian cells ([ILW11]).
- Stratified variable fiveprime end mapping
This multidimensional mapping function behaves as variable fiveprime end mapping, and additionally returns arrays that are stratified by read length.
For each region of interest, a 2D array is returned, in which each row represents a given read length, and each column a nucleotide position in the region of interest. In other words, summing over the columns produces the same array that would be given by variable fiveprime end mapping.
- Threeprime end mapping
Each read alignment is mapped to its 3’ end, or at a fixed offset (in nucleotides) from its 3’ end.
- Entire or Center-weighted mapping
Zero or more positions are trimmed from each end of the read alignment, and the remaining N positions in the alignment are incremented by 1/N read counts (so that each read is still counted once, when integrated over its mapped length).
This is also used for ribosome profiling of E. coli ([OBS+11]) and D. melanogaster ([DFB+13]), and RNA-seq.
In the image below, the same set of read alignments from a ribosome profiling experiment is mapped under various functions. Note the start codon peak and stop codon peak that appear when reads are mapped to specific locations:
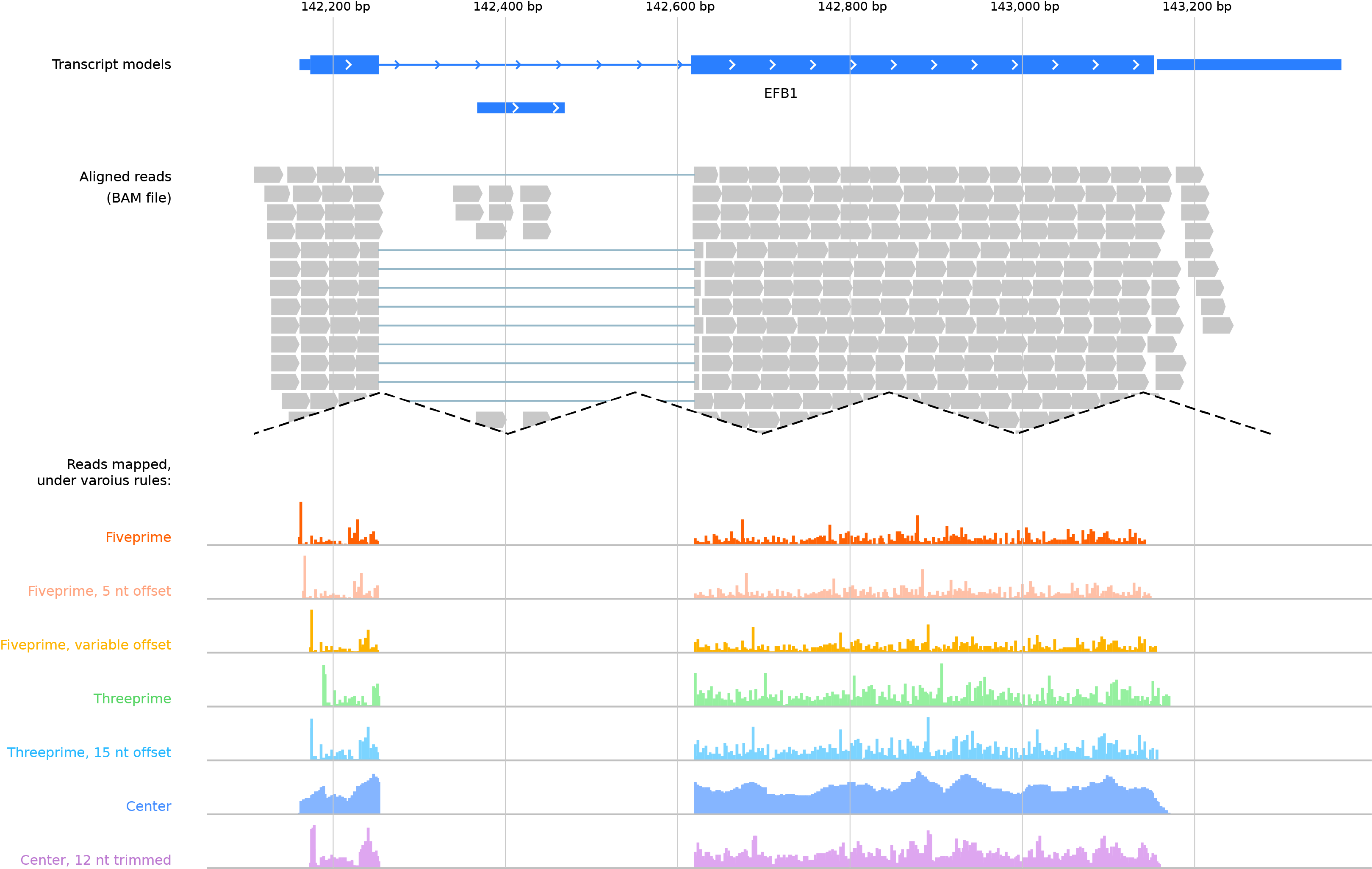
Top: gene model. Middle: alignments of ribosome-protected footprints, displayed as in the IGV genome browser without a mapping function. Bottom rows: Ribosome footprints mapped under various mapping functions.¶
Choosing mapping functions in command-line scripts¶
Mapping functions may be specified to command-line scripts
using the following command-line arguments (note, command-line scripts don’t at
present support mapping functions that return multidimensional arrays):
Mapping function
Argument
Fiveprime
--fiveprimeFiveprime variable
--fiveprime_variableThreeprime
--threeprimeCenter/entire
--center
The following arguments additionally influence how mapping functions behave:
Argument
Behavior
--offset XFor
--fiveprimeor--threeprime,Xis taken to be an integer specifying the offset into the read, at which read alignments should be mapped.For
--fiveprime_variable,Xis taken to be the filename of a two-column tab-delimited text file, in which first column represents read length or the special keyword ‘default’, and the second column represents the offset from the five prime end at which reads of that length should be mapped.
--nibble X
Xis taken to be the number of bases to trim from each end of the read before mapping.
See the documentation for individual command-line scripts
for a detailed discussion of their arguments.
Choosing mapping functions in interactive Python sessions¶
Mapping functions in plastid are applied when read alignments
are imported. Read alignments are held in data structures called GenomeArrays
(see plastid.genomics.genome_array).
Alignments in BAM format can be imported into a BAMGenomeArray. Mapping
functions are set via
set_mapping():
>>> from plastid.genomics.genome_array import BAMGenomeArray, FivePrimeMapFactory, CenterMapFactory
>>> alignments = BAMGenomeArray("SRR609197_riboprofile_5hr_rep1.bam")
>>> # map reads 5 nucleotides downstream from their 5' ends
>>> alignments.set_mapping(FivePrimeMapFactory(offset=5))
and, the mapping function for a BAMGenomeArray can be changed at any time:
>>> # map reads along entire lengths
>>> alignments.set_mapping(CenterMapFactory())
Alignments in bowtie format can be imported into a GenomeArray. Because
bowtie files are not sorted or indexed, mapping functions must be applied
upon import, and cannot be changed afterwards:
>>> from plastid.genomics.genome_array import GenomeArray, five_prime_map
>>> # map reads 5 nucleotides downstream from their 5' ends
>>> fiveprime_alignments = GenomeArray()
>>> fiveprime_alignments.add_from_bowtie(open("some_file.bowtie"),five_prime_map,offset=5)
>>> # map reads along entire lengths
>>> entire_alignments = GenomeArray()
>>> entire_alignments.add_from_bowtie(open("some_file.bowtie"),center_map)
Method names for the various mapping functions appear below:
Mapping function |
||
Fiveprime |
||
Fiveprime variable |
||
Stratified fiveprime variable |
not implemented |
|
Threeprime |
||
Center/entire |
Writing new mapping functions¶
Mapping functions in plastid are implemented as callables (functions or
callable class instances). Mapping functions for BAMGenomeArray require the
following signatures:
Parameters¶
- alignments
A list of read alignments represented as
pysam.AlignedSegmentobjects. These correspond to the alignments that will be mapped. Typically, these overlap segment.- segment
A
GenomicSegmentcorresponding to a region of interest
Return values¶
- list
A list of read alignments (
pysam.AlignedSegment) that map within segment under the mapping function implemented by the function.numpy.ndarrayAn array of values at each position in segment, from left-to-right / lowest-to-highest coordinates relative to the genome (not relative to the segment).
More generally, if the mapping function returns a multi-dimensional array, the last dimension must represent the positions in segment (e.g., for a 2D array, the columns would represent the nucleotide positions).
Example 1: Fiveprime alignment mapping¶
This mapping function maps read alignments to their 5’ ends, allowing an optional offset:
>>> import numpy
>>> import warnings
>>> def fiveprime_map_function(alignments,segment,offset=0):
>>> reads_out = []
>>> count_array = numpy.zeros(len(segment))
>>> for read in alignments:
>>> if offset > len(read.positions):
>>> warnings.warn("Offset %snt greater than read length %snt. Ignoring." % (offset,len(read)),
>>> UserWarning)
>>> continue # skip read if offset is outside read boundaries
>>>
>>> # count offset 5' to 3' if the `segment` is on the plus-strand
>>> # or is unstranded
>>> if segment.strand == in ("+","."):
>>> p_site = read.positions[offset]
>>> # count offset from other end if `segment` is on the minus-strand
>>> else:
>>> p_site = read.positions[-offset - 1]
>>>
>>> if p_site >= segment.start and p_site < segment.end:
>>> reads_out.append(read)
>>> count_array[p_site - seg.start] += 1
>>>
>>> return reads_out, count_array
But, BAMGenomeArray will only pass the parameters alignments and segment
to mapping functions. To specify an offset, use a wrapper function:
>>> def MyFivePrimeMapFactory(offset=0):
>>> def new_func(alignments,segment):
>>> return fiveprime_map_function(alignments,segment,offset=offset)
>>>
>>> return new_func
>>> alignments = BAMGenomeArray("SRR609197_riboprofile_5hr_rep1.bam")
>>> alignments.set_mapping(MyFivePrimeMapFactory(offset=5))
Example 2: mapping alignments to their mismatches¶
BAM files contain rich information about read alignments, and these are
exposed to us via pysam.AlignedSegment. This mapping function maps
read alignments to sites where they mismatch a reference genome,
assuming the alignment contains no indels. Mismatch information is pulled from
the MD tag for each read alignment:
>>> import re
>>> nucleotides = re.compile(r"[ACTGN]")
>>>
>>> def mismatch_mapping_function(alignments,segment):
>>> reads_out = []
>>> count_array = numpy.zeros(len(segment))
>>> for read in alignments:
>>> for tag,val in read.tags:
>>> # we are also assuming no indels, which would make parsing MD more complicated.
>>> #
>>> # mismatches are in stored in `MD` tag of reach alignment in SAM/BAM files
>>> # for see MD tag structure http://samtools.sourceforge.net/SAM1.pdf
>>> # they basically look like numbers of matches separated by
>>> # the letter that mismatches. e.g. 12A15C22
>>> # means: 12 matches, followed by mismatch 'A', followed by 15 matches,
>>> # followed by mismatch 'C', followed by 22 matches
>>> #
>>> # convert MD tag to a vector of positions that mismatch
>>> if tag == "MD":
>>> mismatched_positions = numpy.array([int(X) for X in re.split(nucleotides,val)[:-1]])
>>> mismatched_positions += numpy.arange(len(mismatched_positions))
>>>
>>> # figure out coordinate of mismatch with respect to genome and `segment`
>>> for pos in mismatched_positions:
>>> genome_position = read.positions[pos]
>>> segment_position = genome_position - segment.start
>>> count_array[segment_position] += 1
>>>
>>> return reads_out, count_array
This mapping function may then be used as above:
>>> alignments.set_mapping(mismatch_mapping_function)
See also¶
Extending plastid for details on making custom mapping functions accessible to command-line scripts
P-site mapping example, in which a mapping function for ribosome profiling data is derived and applied
Module documentation for
plastid.genomics.map_factoriesandplastid.genomics.genome_array, which provide more details on mapping functions,BAMGenomeArrays, andGenomeArrays